EMSL Arrows - Evolution of Chemical and Materials Computation¶
Overview¶
We would like thank the DOD SERDP program and the DOE OS OBER EMSL project for providing support that helped with the initial development of EMSL Arrows.
Tutorial on YouTube (mobile devices)
Click here to try out Arrows by sending it an email
Are you just learning NWChem and would like to have an easy way to generate input decks, check your output decks against a large database of calculations, perform simple thermochemistry calculations, calculate the NMR and IR spectra of modest size molecule, or just try out NWChem before installing it? EMSL Arrows scientific service can help. A Web API to EMSL Arrows is now available for Alpha testing. Click on this link.
For more information contact Eric Bylaska (eric.bylaska@pnnl.gov)
The difficulty of simulating the thermodynamic and kinetic properties of new materials is convoluted by the sensitivity of the processes at the macroscopic scale to the atomic scale; the unusual and unexpected bonding behaviors of the materials; the complex extreme temperature and pressure environments likely to be encountered; and the requirements that simulations be as parameter free as possible and extremely reliable. The tools of quantum chemistry and statistical mechanics combined with advanced parallel packages such as NWChem have proved to be very effective and productive. Not surprisingly, programs that implement these types of tools make up a large fraction of DOE OS supercomputer cycles. Despite these hugely successful theoretical developments, reliable calculations of this type require considerable computational effort and often the use of codes with difficult input decks.
The NWChem molecular modeling software implements a robust and diverse set of molecular theories that can estimate the thermodynamics and kinetics of molecules and materials. It arguably has the most capabilities of any molecular modeling code today. The problem with NWChem and other molecular modeling codes is that:
- Molecular modeling software is extremely complex, contains millions of lines of code, and takes a long time to set up and to learn how to use.
- Even the most basic input for molecular modeling software requires the use of other software to generate it.
- Because of this complexity people unnaturally identify with codes and molecular theories, and they are hesitant to learn new codes and new molecular simulation techniques.
The goal of this project is to provide EMSL users and DOE scientists and engineers with an open-source computational chemistry and materials tool called EMSL Arrows. EMSL Arrows is a software package that combines NWChem, SQL and NOSQL databases, and email (in the future also social networks, e.g. Twitter, Tumblr) that simplifies molecular and materials modeling and makes these modeling capabilities easier to use and more accessible to many scientists and engineers.
EMSL Arrows is very simple to use. The user just emails chemical reactions to arrows@emsl.pnnl.gov and then an email is sent back with thermodynamic, reaction pathway (kinetic), spectroscopy, and other results.
EMSL Arrows parses the email and then searches the database for the compounds in the reactions. If a compound isn’t there, an NWChem calculation is setup and submitted to calculate it. Once the calculation is finished the results are entered into the database and then results are emailed back. This whole process is completely automated. To enter different calculation types (e.g. use pspw theory, or pbe0 exchange correlation functional) the SMILES is appended with keyword{options} tags. An example email is as follows:
To: arrows@emsl.pnnl.gov
Subject: Calculate isodesmic reactions
Arrows::
Reaction: C(Cl)(Cl)(Cl)O + C --> C(Cl)(Cl)Cl + CO :Reaction
Reaction: C(Cl)(Cl)(Cl)O + C --> C(Cl)(Cl)Cl + CO ~ theory{pspw} :Reaction
Reaction: C(Cl)(Cl)(Cl)S + C --> C(Cl)(Cl)Cl + CS :Reaction
Reaction: C(Cl)(Cl)(Cl)S + C --> C(Cl)(Cl)Cl + CS ~ theory{pm3} :Reaction
Reaction: TNT + 3 benzene --> toluene + 3 nitrobenzene ~ xc{pbe} :Reaction
::Arrows
The results returned by EMSL Arrows are a combination of text and graphical output.
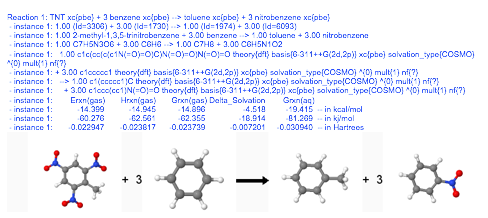
Currently EMSL Arrows is designed to calculate the following for all NWChem theories:
- Reaction thermodynamics for molecular systems
- Reaction paths for molecular systems
- UV-vis, IR, Raman spectra for molecular systems, phonon spectra for materials systems
- NMR spectra for molecular and materials systems
- EXAFS spectra for molecular and materials systems
- Energetics, structures, and band structures of crystals using the Crystal Open Database (COD ) numbers
- A variety of datafiles can be returned including XYZ files, CIF files, NWChem output files
We envision that as Arrows evolves it will be part of future closed cycles of chemical and materials discovery that requires integrated computational and experimental tools combined with materials synthesis.
Try out EMSL Arrows by sending the following simple emails to arrows@emsl.pnnl.gov¶
Returns b3lyp/6-311++G(2d,2p) results for the cinnamon flavored molecule. Click here to run this example.
---------------- mailto: arrows@emsl.pnnl.gov -----------------------
Arrows::
molecule: Cinnamaldehyde :molecule
::Arrows
Using MP2 to calculate the reaction energy of a hydrolysis reaction for TNT. Click here to run this example.
{kind=link}
---------------- mailto: arrows@emsl.pnnl.gov -----------------------
Arrows::
Reaction: cid=8376 + hydroxide --> O=N(=O)c1cc(O)c(c(c1)N(=O)=O)C + nitrite ~ theory{mp2} :Reaction
::Arrows
Examples of isodesmic reaction Click here to to run this example.
---------------- mailto: arrows@emsl.pnnl.gov -----------------------
Arrows::
Reaction: TNT + 3 benzene --> toluene + 3 nitrobenzene ~ theory{mp2} :Reaction
Reaction: C(Cl)(Cl)(Cl)O + C --> C(Cl)(Cl)Cl + CO :Reaction
Reaction: C(Cl)(Cl)(Cl)O + C --> C(Cl)(Cl)Cl + CO ~ xc{pbe} :Reaction
Reaction: C(Cl)(Cl)(Cl)O + C --> C(Cl)(Cl)Cl + CO ~ theory{pspw} :Reaction
::Arrows
Examples of reaction prediction capabilities in Arrows. Click here to to run this example.
---------------- mailto: arrows@emsl.pnnl.gov -----------------------
Arrows::
Predict: 2 methane :Predict
::Arrows
Click here to to run this example.
---------------- mailto: arrows@emsl.pnnl.gov -----------------------
Arrows::
Predict: TNT + hydroxide :Predict
::Arrows
Fetch an NWChem output deck from Arrows. Click here to to run this example.
---------------- mailto: arrows@emsl.pnnl.gov -----------------------
Arrows::
nwoutput: caffeine :nwoutput
::Arrows
Try out the following web API links (Now Available for Alpha Testing)¶
Introduction to ESMILES - How to Change Calculation Theories¶
The combined string, Molecule_Input keyword1{option1}
keyword2{option2} keywordN{optionN}, is called an extended smiles or
esmiles for short. The Molecule_Input can be specified using a
variety of formats including a SMILES string, common names, iupac, kegg
numbers, cas, pubchem ids, chemspider ids, and InChI strings. The
keyword{option} tags are used to enter different calculation types for a
molecule, e.g. use pspw theory, ccsd(t), or pbe0 exchange correlation
functional.
The following are examples of esmiles strings:
Plane-Wave DFT calculation using LDA and a cutoff energy=30.0 Ry
c1ccccc1 theory{pspw} xc{lda} basis{30.0 Ry}
MP2 calculation using 6-31G* basis set
CCO theory{mp2} basis{6-31G*}
CCSD(T) calculation of ethanol
CCO theory{ccsd(t)} basis{6-31G*}
Mopac PM3 calculation of caffeine
Caffeine theory{pm3}
Aperiodic plane-wave DFT calculation of triplet cabon tetrachloride
C(Cl)(Cl)(Cl)Cl mult{3} theory{pspw4}
Gas-phase M06-2x/6-31+G* calculation of benzene
benzene theory{dft} xc{m06-2x} solvation_type{none}
Equivalent ESMILES for CCSD(T)/6-31G* calculation of methanol
methyl alcohol theory{ccsd(t)} basis{6-31G*}
kegg=D02309 theory{ccsd(t)} basis{6-31G*}
cas=67-56-1 theory{ccsd(t)} basis{6-31G*}
cid=887 theory{ccsd(t)} basis{6-31G*}
csid=864 theory{ccsd(t)} basis{6-31G*}
InChI=1S/CH4O/c1-2/h2H,1H3 theory{ccsd(t)} basis{6-31G*}
The available keywords in and esmiles string are: theory, theory_property, theory_base, basis, basis_property, basis_base, xc, xc_property, xc_base, solvation_type, charge, mult, xyzdata, geometry_generation, and calculation_type.
ESMILES Options - theory{}, theory_property{} and theory_base{}¶
The default theory used is theory{dft}. The following theories are available:
- dft -- NWChem Gaussian DFT
- pspw -- NWChem Plane-Wave DFT (periodic boundary conditions, Γ point)
- pspw4 -- NWChem Plane-Wave DFT (aperiodic boundary conditions)
- mp2 -- NWChem MP2 program
- ccsd(t) -- NWChem CCSD(T)
- pm3 -- Mopac7 PM3
- am1 -- Mopac7 AM1
- mindo -- Mopac7 MINDO
- mindo3 -- Mopac7 MINDO3
The theory_property{} is an optional keyword used to specify the theory used in an nmr calculation, and theory_base{} is an optional keyword used to specify the theory of the base calculation for an MP2 or CCSD(T) calculation. By default the theory_property and theory_base are defined to be the same as theory{}.
ESMILES Options - basis{}, basis_property{} and basis_base{}¶
The default basis used is 6-311++G(2d,2p) for the Gaussian DFT, MP2 and CCSD(T) programs. For plane-wave DFT the default basis or cutoff energy is defined to by 50.0 Hartrees or 100.0 Ry.
For Gaussian basis sets any basis set recognized by NWChem can be used, e.g.
CCO basis{6-31G*}
Other common basis sets can be used such as cc-pvdz, 6-311G, 3-21G, 6-31+G*.
For plane-wave basis sets the cutoff energy can changed by just entering the number in Hartrees or Rydbergs
CCO theory{pspw] basis{50.0}
CCO theory{pspw} basis{100 Ry}
The basis_property{} is an optional keyword used to specify the basis set used in an nmr calculation, and basis_base{} is an optional keyword used to specify the basis set of the base calculation for an MP2 or CCSD(T) calculation. By default the basis_property and basis_base are defined to be the same as basis{}.
ESMILES Options - xc{}, xc_property{} and xc_base{}¶
Only the Gaussian and plane-wave DFT programs utilize the xc{} keyword. The default exchange correlation functional used is xc{b3lyp}. The following exchange correlation functions are available with the Gaussian DFT and plane-wave DFT programs.
- lda -- local density approximation (LDA) of S.J. Vosko, L. Wilk and M. Nusair, Can. J. Phys. 58, 1200 (1980)
- pbe -- The gradient corrected exchange correlation function of J.P. Perdew, K. Burke and M. Ernzerhof,
Phys. Rev. Lett. 77, 3865 (1996); 78 , 1396 (1997)
- blyp -- The gradient corrected exchange correlation function A.D. Becke, Phys. Rev. A 88, 3098 (1988) and
C. Lee, W. Yang and R. G. Parr, Phys. Rev. B 37, 785 (1988)
- b3lyp -- the hybrid exchange correlation function of A.D. Becke, J. Chem. Phys. 98, 5648 (1993)
and C. Lee, W. Yang and R. G. Parr, Phys. Rev. B 37, 785 (1988)
- pbe0 -- the hybrid exchange correlation function of C.Adamo and V.Barone, J. Chem. Phys. 110, 6158 (1999)
- m06-2x -- the hybrid meta exchange correlation function of Y. Zhao, D. G. Truhlar, J. Chem. Phys. 125, 194101 (2006).
Only available in Gaussian DFT program
The xc_property{} is an optional keyword used to specify the exchange
correlation potential used in an nmr calculation, and xc_base{} is an
optional keyword used to specify the exchange correlation potential of
the base calculation for an MP2 or CCSD(T) calculation. By default the
xc_property and xc_base are defined to be the same as xc{}.
ESMILES Options - solvation_type{}¶
The default solvation type is solvation_type{COSMO}. The following solvation types are available with the Gaussian DFT, MP2 and CCSD(T) programs.
- COSMO -- The COSMO solvation model of Klampt and Shuurman (solvent=water)
- COSMO-SMD -- The extended Minnesota COSMO solvation model of Cramer et al. (solvent=water)
- COSMO-SMD:solvent -- where the solvent keyword is from Table of SMD solvent names below
- None -- Gas-phase calculation, no solvation model included in the calculations
The available SMD solvent keywords are given below:
| Keyword | Name |
|---|---|
| h2o | water (default) |
| water | water (default) |
| acetacid | acetic acid |
| acetone | acetone |
| acetntrl | acetonitrile |
| acetphen | acetophenone |
| aniline | aniline |
| anisole | anisole |
| benzaldh | benzaldehyde |
| benzene | benzene |
| benzntrl | benzonitrile |
| benzylcl | benzyl chloride |
| brisobut | 1-bromo-2-methylpropane |
| brbenzen | bromobenzene |
| brethane | bromoethane |
| bromform | bromoform |
| broctane | 1-bromooctane |
| brpentan | 1-bromopentane |
| brpropa2 | 2-bromopropane |
| brpropan | 1-bromopropane |
| butanal | butanal |
| butacid | butanoic acid |
| butanol | 1-butanol |
| butanol2 | 2-butanol |
| butanone | butanone |
| butantrl | butanonitrile |
| butile | butyl acetate |
| nba | butylamine |
| nbutbenz | n-butylbenzene |
| sbutbenz | sec-butylbenzene |
| tbutbenz | tert-butylbenzene |
| cs2 | carbon disulfide |
| carbntet | carbon tetrachloride |
| clbenzen | chlorobenzene |
| secbutcl | sec-butyl chloride |
| chcl3 | chloroform |
| clhexane | 1-chlorohexane |
| clpentan | 1-chloropentane |
| clpropan | 1-chloropropane |
| ocltolue | o-chlorotoluene |
| m-cresol | m-cresol |
| o-cresol | o-cresol |
| cychexan | cyclohexane |
| cychexon | cyclohexanone |
| cycpentn | cyclopentane |
| cycpntol | cyclopentanol |
| cycpnton | cyclopentanone |
| declncis | cis-decalin |
| declntra | trans-decalin |
| declnmix | decalin (cis/trans mixture) |
| decane | n-decane |
| decanol | 1-decanol |
| edb12 | 1,2-dibromoethane |
| dibrmetn | dibromomethane |
| butyleth | dibutyl ether |
| odiclbnz | o-dichlorobenzene |
| edc12 | 1,2-dichloroethane |
| c12dce | cis-dichloroethylene |
| t12dce | trans-dichloroethylene |
| dcm | dichloromethane |
| ether | diethyl ether |
| et2s | diethyl sulfide |
| dietamin | diethylamine |
| mi | diiodomethane |
| dipe | diisopropyl ether |
| dmds | dimethyl disulfide |
| dmso | dimethyl sulfoxide |
| dma | N,N-dimethylacetamide |
| cisdmchx | cis-1,2-dimethylcyclohexane |
| dmf | N,N-dimethylformamide |
| dmepen24 | 2,4-dimethylpentane |
| dmepyr24 | 2,4-dimethylpyridine |
| dmepyr26 | 2,6-dimethylpyridine |
| dioxane | 1,4-dioxane |
| phoph | diphenyl ether |
| dproamin | dipropylamine |
| dodecan | n-dodecane |
| meg | 1,2-ethanediol |
| etsh | ethanethiol |
| ethanol | ethanol |
| etoac | ethyl acetate |
| etome | ethyl formate |
| eb | ethylbenzene |
| phenetol | ethyl phenyl ether |
| c6h5f | fluorobenzene |
| foctane | 1-fluorooctane |
| formamid | formamide |
| formacid | formic acid |
| heptane | n-heptane |
| heptanol | 1-heptanol |
| heptnon2 | 2-heptanone |
| heptnon4 | 4-heptanone |
| hexadecn | n-hexadecane |
| hexane | n-hexane |
| hexnacid | hexanoic acid |
| hexanol | 1-hexanol |
| hexanon2 | 2-hexanone |
| hexene | 1-hexene |
| hexyne | 1-hexyne |
| c6h5i | iodobenzene |
| iobutane | 1-iodobutane |
| c2h5i | iodoethane |
| iohexdec | 1-iodohexadecane |
| ch3i | iodomethane |
| iopentan | 1-iodopentane |
| iopropan | 1-iodopropane |
| cumene | isopropylbenzene |
| p-cymene | p-isopropyltoluene |
| mesityln | mesitylene |
| methanol | methanol |
| egme | 2-methoxyethanol |
| meacetat | methyl acetate |
| mebnzate | methyl benzoate |
| mebutate | methyl butanoate |
| meformat | methyl formate |
| mibk | 4-methyl-2-pentanone |
| mepropyl | methyl propanoate |
| isobutol | 2-methyl-1-propanol |
| terbutol | 2-methyl-2-propanol |
| nmeaniln | N-methylaniline |
| mecychex | methylcyclohexane |
| nmfmixtr | N-methylformamide (E/Z mixture) |
| isohexan | 2-methylpentane |
| mepyrid2 | 2-methylpyridine |
| mepyrid3 | 3-methylpyridine |
| mepyrid4 | 4-methylpyridine |
| c6h5no2 | nitrobenzene |
| c2h5no2 | nitroethane |
| ch3no2 | nitromethane |
| ntrprop1 | 1-nitropropane |
| ntrprop2 | 2-nitropropane |
| ontrtolu | o-nitrotoluene |
| nonane | n-nonane |
| nonanol | 1-nonanol |
| nonanone | 5-nonanone |
| octane | n-octane |
| octanol | 1-octanol |
| octanon2 | 2-octanone |
| pentdecn | n-pentadecane |
| pentanal | pentanal |
| npentane | n-pentane |
| pentacid | pentanoic acid |
| pentanol | 1-pentanol |
| pentnon2 | 2-pentanone |
| pentnon3 | 3-pentanone |
| pentene | 1-pentene |
| e2penten | E-2-pentene |
| pentacet | pentyl acetate |
| pentamin | pentylamine |
| pfb | perfluorobenzene |
| benzalcl | phenylmethanol |
| propanal | propanal |
| propacid | propanoic acid |
| propanol | 1-propanol |
| propnol2 | 2-propanol |
| propntrl | propanonitrile |
| propenol | 2-propen-1-ol |
| propacet | propyl acetate |
| propamin | propylamine |
| pyridine | pyridine |
| c2cl4 | tetrachloroethene |
| thf | tetrahydrofuran |
| sulfolan | tetrahydrothiophene-S,S-dioxide |
| tetralin | tetralin |
| thiophen | thiophene |
| phsh | thiophenol |
| toluene | toluene |
| tbp | tributyl phosphate |
| tca111 | 1,1,1-trichloroethane |
| tca112 | 1,1,2-trichloroethane |
| tce | trichloroethene |
| et3n | triethylamine |
| tfe222 | 2,2,2-trifluoroethanol |
| tmben124 | 1,2,4-trimethylbenzene |
| isoctane | 2,2,4-trimethylpentane |
| undecane | n-undecane |
| m-xylene | m-xylene |
| o-xylene | o-xylene |
| p-xylene | p-xylene |
| xylenemx | xylene (mixture) |
When a solvent is specified by name, the descriptors for the solvent are based on the Minnesota Solvent Descriptor Database:
Winget, P.; Dolney, D. M.; Giesen, D. J.; Cramer, C. J.; Truhlar, D. G. Minnesota Solvent Descriptor Database. University of Minnesota: Minneapolis, MN, 2010. http://comp.chem.umn.edu/solvation/mnsddb.pdf
ESMILES Reactions - How to Calculate Reaction Energies¶
The basic input is a chemical reaction where the molecules are specified using smiles strings or esmiles strings (vida infra), e.g.
C(Cl)(Cl)(Cl)O + C --> C(Cl)(Cl)Cl + CO
Note that the reaction: :reaction keywords have only one “:”, whereas the Arrows keywords use two colons.
The results contain both gas phase and solution phase reaction energies. The default level of theory used in these calculations is b3lyp/6-311++G(2d,2p) and the default solvation model is COSMO. The returned email will contain the following output.
Reaction 1: C(Cl)(Cl)(Cl)O + C --> C(Cl)(Cl)Cl + CO
- instance 1: 1.00 (Id=6833) + 1.00 (Id=11824) --> 1.00 (Id=6832) + 1.00 (Id=11215)
- instance 1: 1.00 trichloromethanol + 1.00 methane --> 1.00 chloroform + 1.00 methanol
- instance 1: 1.00 C1Cl3H1O1 + 1.00 C1H4 --> 1.00 C1Cl3H1 + 1.00 C1H4O1
- instance 1: 1.00 OC(Cl)(Cl)Cl theory{dft} basis{6-311++G(2d,2p)} xc{b3lyp} solvation_type{COSMO} ^{0} mult{1} nf{?}
- instance 1: + 1.00 C theory{dft} basis{6-311++G(2d,2p)} xc{b3lyp} solvation_type{COSMO} ^{0} mult{1} nf{0}
- instance 1: --> 1.00 C(Cl)(Cl)Cl theory{dft} basis{6-311++G(2d,2p)} xc{b3lyp} solvation_type{COSMO} ^{0} mult{1} nf{?}
- instance 1: + 1.00 CO theory{dft} basis{6-311++G(2d,2p)} xc{b3lyp} solvation_type{COSMO} ^{0} mult{1} nf{0}
- instance 1: Erxn(gas) Hrxn(gas) Grxn(gas) Delta_Solvation Grxn(aq)
- instance 1: 8.035 9.580 8.809 -1.991 6.818 -- in kcal/mol
- instance 1: 33.618 40.084 36.857 -8.332 28.525 -- in kj/mol
- instance 1: 0.012804 0.015267 0.014038 -0.003173 0.010865 -- in Hartrees
The reaction output for the chemical reaction contains the gas phase reaction energy, gas-phase reaction enthalpy, gas-phase reaction free energy, change in solvation energy, and the solution phase reaction free energy. The energy values are given in kcal/mol, kj/mol, and Hartrees.. Besides the energies the output also provides several rows of information about the calculation:
- first row: the reaction input parsed
- second row: the arrows ids used for the compounds in the reaction
- third row: the iupac names of the compounds if available. If not available the systems will default to using smiles
strings
- fourth- rows: the chemical reaction is written using the esmiles notation.
The esmiles notation contains all the information about the calculations of the compounds. In this example, theory used was dft, basis was 6-311++G(2d,2p), the exchange correlation, the solvation type was cosmo. The charge and multiplicity of the molecules are also given. The value in the nf{} tag contains the number of imaginary frequencies in the vibrational calculation for the molecule.
A variety of other inputs to describe the chemical structure besides smiles can be used, including common names, iupac, kegg numbers, cas, pubchem ids, chemspider ids, and InChI strings. The common names, iupac and InChI strings are entered as replacements to the smiles strings, and the kegg, cas, pubchem, and csid inputs are entered as kegg=value, cas=value, cid=value, csid=value where value is the id. The chemical structure input types can be mixed and matched in the reaction input. The following reaction inputs are all equivalent.
trichloromethanol + methane --> chloroform + methyl alcohol
trichloromethanol + C --> chloroform + kegg=D02309
trichloromethanol + C --> chloroform + cas=67-56-1
trichloromethanol + C --> chloroform + cid=887
trichloromethanol + C --> chloroform + csid=864
trichloromethanol + C --> chloroform + InChI=1S/CH4O/c1-2/h2H,1H3
To calculate atomization energies the following input can be used.
C(Cl)(Cl)(Cl)O --> [C] mult{3} + 3 [Cl] mult{2} + [O] mult{3}
MAP Function for Adding Options to Reactions¶
To calculate a reaction energy using non-default options the following format could be used, e.g.
Arrows::
reaction:
trichloromethanol theory{pspw} xc{lda} + methane theory{pspw} xc{lda}
--> chloroform theory{pspw} xc{lda} + methyl alcohol theory{pspw} xc{lda}
:reaction
::Arrows
in the body of an Arrows email, or just the following single line input in the Web API entry box
trichloromethanol theory{pspw} xc{lda} + methane theory{pspw} xc{lda}
--> chloroform theory{pspw} xc{lda} + methyl alcohol theory{pspw} xc{lda}
Entering ESMILES in this way for reactions is tedius and prone to typos. To simplify this type of input a map function has been added to the reaction input, where the format for the mapping function is to append the reaction with the tilde, “~“, symbol followed by the esmiles options.
trichloromethanol + methane --> chloroform + methyl alcohol ~ theory{pspw} xc{lda}
The map function essentially appends every compound in the reaction by
the esmiles options string.This is preferred way to use the map
function. However, an alternative format for entering the map function
has also been added to the reaction: :reaction block. The format of the
block is reaction[esmiles options]: reaction :reaction.
Arrows::
reaction[theory{pspw} xc{lda}]:
trichloromethanol + methane --> chloroform + methyl alcohol
:reaction
::Arrows
How to Define the Chemical Structure with XYZ Input¶
The xyzinput: :xyzinput block is used to enter a chemical structure using xyz coordinates. The label: :label subblock is used to label the xyz structure so that it can be referenced in reaction: :reaction, molecule: :molecule, and nmr: :nmr blocks. The xyz geometry is entered inside the xyzdata: :xyzdata block. The coordinates are assumed to be in Angstroms. The xyz geometry can either contain the number of atoms at the start of the input, e.g.
Arrows::
xyzinput:
label: amolecule :label
xyzdata:
20
C 0.810772 1.260891 0.224768
C -0.445319 0.626551 0.148559
C -0.550132 -0.747571 -0.024182
C 0.598317 -1.510887 -0.051277
C 1.856720 -0.927387 0.081993
C 1.951003 0.440481 0.208335
H 2.736961 -1.550133 0.062422
H 2.912395 0.927722 0.273890
O 1.062201 2.575051 0.296009
C 0.213380 3.557631 -0.323370
H -1.520657 -1.209783 -0.105115
N -1.712300 1.341956 0.351481
N 0.485785 -2.966232 -0.210786
O -0.636770 -3.441145 -0.327238
O 1.526277 -3.613525 -0.218259
O -2.671572 1.004073 -0.327713
O -1.733900 2.198527 1.228109
H 0.882435 4.349335 -0.647148
H -0.510291 3.940088 0.389177
H -0.297779 3.136834 -1.188838
:xyzdata
:xyzinput
molecule: label=amolecule xc{m06-2x} :molecule
::Arrows
r it can be left out, e.g.
Arrows::
xyzinput:
label: amolecule :label
xyzdata:
C 0.810772 1.260891 0.224768
C -0.445319 0.626551 0.148559
C -0.550132 -0.747571 -0.024182
C 0.598317 -1.510887 -0.051277
C 1.856720 -0.927387 0.081993
C 1.951003 0.440481 0.208335
H 2.736961 -1.550133 0.062422
H 2.912395 0.927722 0.273890
O 1.062201 2.575051 0.296009
C 0.213380 3.557631 -0.323370
H -1.520657 -1.209783 -0.105115
N -1.712300 1.341956 0.351481
N 0.485785 -2.966232 -0.210786
O -0.636770 -3.441145 -0.327238
O 1.526277 -3.613525 -0.218259
O -2.671572 1.004073 -0.327713
O -1.733900 2.198527 1.228109
H 0.882435 4.349335 -0.647148
H -0.510291 3.940088 0.389177
H -0.297779 3.136834 -1.188838
:xyzdata
:xyzinput
molecule: label=amolecule xc{m06-2x} :molecule
::Arrows
How to Calculate NMR Spectra¶
The nmr: :nmr block is used to energy an NMR calculation
Arrows::
nmr: c1ccccc1 basis{6-31G*} solvation_type{None} :nmr
::Arrows
For single line input the esmiles is preceded by the words “nmr for”, e.g.
nmr for c1ccccc1 basis{6-31G*} solvation_type{None}
How to Generate a Table of Reactions¶
The reactionenumerate: :reactionenumerate block is used to generate a table of reactions in CSV format, which can be copy and pasted into spreadsheets.
Arrows::
reactionenumerate:
energytype: grxn(aq) kcal/mol :energytype
tablereactions:
reaction: TNT + hydroxide --> TNT-2-OH + nitrite :reaction
reaction: DNAN + hydroxide --> DNAN-2-OH + nitrite :reaction
:tablereactions
tablemethods:
method: xc{pbe} :method
method: xc{b3lyp} :method
method: xc{m06-2x} :method
:tablemethods
:reactionenumerate
::Arrows
How to Fetch NWChem Output¶
The NWChem output can be fetched using the nwoutput: :nwoutput and printnwout: :printnwout blocks. The input for the nwoutput: :nwoutput block is an ESMILES strings, e.g.
Arrows::
nwoutput: TNT theory{pspw} :nwoutput
::Arrows
For single line input the esmiles is preceded by the words “nwoutput for”, e.g.
nwoutput for aspirin theory{pspw}
The input for the printnwout: :printnwout block is an Arrows id, e.g.
Arrows::
printnwout: 13212 :printnwout
::Arrows
Generate NWChem Input¶
The Web API can be used to generate an NWChem input deck. For single line input the esmiles is preceded by the words “input deck for”, e.g.
input deck for aspirin
How to Fetch XYZ Geometry¶
An XYZ geometry can be fetched using the xyzfile: :xyzfile and printxyz: :printxyz blocks. The input for the xyzfile: :xyzfile block is an ESMILES strings, e.g.
Arrows::
xyzfile: TNT theory{pspw} :xyzfile
::Arrows
The input for the printxyz: :printxyz block is an Arrows id, e.g.
Arrows::
printxyz: 13212 :printxyz
::Arrows
For single line input the esmiles is preceded by the words “xyz for”, e.g.
xyz for TNT theory{pspw}