Properties¶
Overview¶
Properties can be calculated for both the Hartree-Fock and DFT wave functions. The properties that are available are:
- Natural bond analysis
- Dipole, quadrupole, and octupole moment
- Mulliken population analysis and bond order analysis
- Electrostatic potential (diamagnetic shielding) at nuclei
- Electric field and field gradient at nuclei
- Electric field gradients with relativistic effects
- Electron and spin density at nuclei
- NMR shielding (GIAO method)
- NMR hyperfine coupling (Fermi-Contact and Spin-Dipole expectation values)
- NMR indirect spin-spin coupling
- Gshift
- Response to electric and magnetic fields (static and dynamic)
- Raman
- Localization of molecular orbitals
The properties module is started when the task directive TASK
PROPERTY
[property keyword]
[CENTER ((com || coc || origin || arb <real x y z>) default coc)]
END
Most of the properties can be computed for Hartree-Fock (closed-shell RHF, open-shell ROHF, and open-shell UHF), and DFT (closed-shell and open-shell spin unrestricted) wavefunctions. The NMR hyperfine and indirect spin-spin coupling require a UHF or ODFT wave function.
Vectors keyword¶
VECTORS [ (<string input_movecs >)]
The VECTORS directive allows the user to specify the input molecular orbital vectors for the property calculation
Property keywords¶
Each property can be requested by defining one of the following keywords:
NBOFILE
DIPOLE
QUADRUPOLE
OCTUPOLE
MULLIKEN
ESP
EFIELD
EFIELDGRAD
EFIELDGRADZ4
GSHIFT
ELECTRONDENSITY
HYPERFINE [<integer> number_of_atoms <integer> atom_list]
SHIELDING [<integer> number_of_atoms <integer> atom_list]
SPINSPIN [<integer> number_of_pairs <integer> pair_list]
RESPONSE [<integer> response_order <real> frequency]
AIMFILE
MOLDENFILE
LOCALIZATION
ALL
The ALL keyword generates all currently available properties.
NMR and EPR¶
Both the NMR shielding and spin-spin coupling have additional optional
parameters that can be defined in the input. For the shielding the user
can define the number of atoms for which the shielding tensor should be
calculated, followed by the list of specific atom centers. In the case
of spin-spin coupling the number of atom pairs, followed by the atom
pairs, can be defined (i.e., spinspin 1 1 2 will calculate the coupling
for one pair, and the coupling will be between atoms 1 and 2).
For both the NMR spin-spin and hyperfine coupling the isotope that has the highest abundance and has spin, will be chosen for each atom under consideration.
Calculating EPR and paramagnetic NMR parameters¶
The following tutorial illustrates how to combine the hyperfine, gshift and shielding to calculate the EPR and paramagnetic NMR parameters of an open-shell system. All calculations are compatible with the ZORA model potential approach.
For theoretical and computational details, please refer to references123.
NMR: Input Example¶
geometry nocenter
C 0.00000000 0.00000000 0.00000000
O 1.18337200 0.00000000 0.00000000
H -.63151821 0.94387462 0.00000000
end
basis
"*" library 6-311G**
end
property
efieldgradz4 1 3
shielding 2 1 2
hyperfine 2 1 3
gshift
end
relativistic
zora on
zora:cutoff_NMR 1d-8
zora:cutoff 1d-30
end
dft
mult 2
xc becke88 perdew86
end
task dft property
Electric field¶
efieldcomputes the electric field.efieldgradcomputes the electric field gradient.efieldgradZ4computes the electric field gradient based on the two-component relativistic zeroth-order regular approximation (ZORA)
The method used in NWChem to computer electric field and electric fieled gradients in described in publication 4.
CENTER: Center of expansion for multipole calculations¶
The user also has the option to choose the center of expansion for the dipole, quadrupole, and octupole calculations.
[CENTER ((com || coc || origin || arb <real x y z>) default coc)]
com is the center of mass, coc is the center of charge, origin is (0.0, 0.0, 0.0) and arb is any arbitrary point which must be accompanied by the coordinated to be used. Currently the x, y, and z coordinates must be given in the same units as UNITS in GEOMETRY.
Response Calculations¶
Response calculations can be calculated as follows:
property
response 1 7.73178E-2 # response order and frequency in Hartree energy units
velocity # use modified velocity gauge for electric dipole
orbeta # calculate optical rotation 'beta' directly [^5]
giao # GIAO optical rotation [^6][^7][^8], forces orbeta
bdtensor # calculates B-tilde of Refs. [^6][^8]
analysis # analyze response in terms of MOs [^8]
damping 0.007 # complex response functions with damping, Ref [^9]
convergence 1e-4 # set CPKS convergence criterion (default 1e-4)
end
Response calculations are currently supported only for
- order 1 (linear response),
- single frequency,
- electric field,
- mixed electric-magnetic field perturbations.
The output consists of the electric polarizability
and optical rotation tensors (alpha, beta for optical rotation) in
atomic units.
The response keyword requires two arguments: response order and frequency in Hartree energy units
(the aoresponse keyword can be used with same effect as the response keyword).
If the velocity or giao keywords are absent, the
dipole-length form will be used for the dipole integrals. This is a bit
faster.
The isotropic optical rotation is origin independent when using
the velocity gauge (by means of velocity keyword) or with GIAOs 6 (by means of the giao keyword).
With the keyword bdtensor, a
fully origin-invariant optical rotation tensor is calculated 68.
Note that velocity and orbeta are incompatible.
The input line
set prop:newaoresp 0
outside of the properties block forces the use of an
older version of the response code, which has fewer features (in
particular, no working GIAO optical rotation) but which has been tested
more thoroughly. In the default newer version you may encounter
undocumented features (bugs).
The keyword analysis triggers an analysis of the
response tensors in terms of molecular orbitals.
If the property input block also contains the keyword pmlocalization, then the analysis is
performed in terms of Pipek-Mezey localized MOs, otherwise the canonical
set is used (this feature may currently not work, please check the sum
of the analysis carefully). See Ref. [6] for an example. Works with HF
and density functionals for which linear response kernels are
implemented in NWChem.
Please refer to papers6571098 for further details:
Raman¶
Raman calculations can be performed by specifying the Raman block. These calculations are performed in conjunction with polarizability calculations. Detailed description of input parameters at https://pubs.acs.org/doi/10.1021/jp411039m#notes-1
RAMAN
[ (NORMAL | | RESONANCE) default NORMAL ]
[ (LORENTZIAN | | GAUSSIAN) default LORENTZIAN ]
[ LOW <double low default 0.0> ]
[ HIGH <double high default highest normal mode> ]
[ FIRST <integer first default 7> ]
[ LAST < integer last default number of normal modes > ]
[ WIDTH <double width default 20.0> ]
[ DQ <double dq default 0.01> ]
END
task dft raman
or
task dft raman numerical
Sample input block:
property
response 1 8.8559E-2
damping 0.007
end
raman
normal
lorentzian
end
Raman Keywords¶
NORMALandRESONANCE: Type of Raman plot to make.LORENTZIANandGAUSSIAN: Generation of smoothed spectra (rather than sticks) using either a Lorentzian function or a Gaussian function. The default isLORENTZIAN.LOWandHIGH: The default range in which to generate the Raman spectrum plot is (0.0, highest wavenumber normal mode) cm-1. TheLOWandHIGHkeywords modify the frequency range.FIRSTandLAST: The default range of indices of normal modes used in the plot is (7, number of normal modes). TheFIRSTandLASTkeywords modify the range of indices.WIDTH: Controls the width in the smoothed peaks, using Lorentzians or Gaussians, in the plot. The default value forWIDTHis 20.0.DQ: Size of the steps along the normal modes. The default value forDQis 0.01. It is related to the step size dR used in numerical evaluation of polarizability derivative
Raman Output¶
Raman spectrum in stick format and smoothed using Lorentzians or
Gaussians stored in a filename with format [fname].normal.
The number of points is 1000 by default. This value can be changed by adding the following SET directive to the input file
set raman:numpts <integer>
Raman References¶
Please refer to papers1112 for further details:
Polarizability computed with the Sum over Orbitals method¶
As an alternative to the linear response method, the Sum over Orbitals (SOO) method is available to compute polarizabilities. Results of these method are much less accurate than linear response calculations, with values off by a factor of 2-4x. However, the qualitative nature of this results can be used to compute Raman frequencies when coupled with QMD, as described in references 1314.
Sample input computing polarizability both with the SOO method and the linear response method:
property
polfromsos
end
task dft property
property
response 1 0
end
task dft property
Nbofile¶
The keyword NBOFILE does not execute the Natural Bond Analysis code, but
simply creates an input file to be used as input to the stand-alone NBO
code. All other properties are calculated upon request.
Following the successful completion of an electronic structure
calculation, a Natural Bond Orbital (NBO) analysis may be carried out by
providing the keyword NBOFILE in the PROPERTY directive. NWChem will
query the rtdb and construct an ASCII file, file_prefix.gen, that may
be used as input to the stand alone version of the NBO program, GenNBO.
file_prefix is equal to string following the START directive. The
input deck may be edited to provide additional options to the NBO
calculation, (see the NBO user’s manual for details.)
Users that have their own NBO version can compile and link the code into the NWChem software. See the INSTALL file in the source for details.
Aimfile¶
This keyword generates AIM Wavefunction files. The resulting AIM wavefunction file (.wfn/.wfx) can be post-processed with a variety of codes, e.g.
WARNING: Since we have discovered issues in generating .WFN files with this module (e.g. systems with ECPs), the recommended method for generating .WFN file is to first generate a Molden file with the Moldenfile option, then convert the Molden file into a WFN file by using the Molden2AIM program.
Moldenfile¶
MOLDENFILE
MOLDEN_NORM (JANPA | | NWCHEM || NONE)
This keyword generates files using the Molden format. The resulting Molden file (.molden) should compatible with a variety of codes that can input Molden files, e.g.
- Molden
- JANPA (the nwchem2molden step is no longer
required when using .molden files and the
MOLDEN_NORM JANPAkeyword) - orbkit
- Molden2qmc
- Molden2AIM
- Multiwfn
the MOLDEN_NORM option allows the renormalization of the basis set
coefficients. By default, the coefficient values from input are not
modified. Using the JANPA value coefficients are normalized following
JANPA’s
convention (where basis coefficients are normalized to unity), while the NWCHEM will produce coefficients normalized
according to NWChem’s convention. Using MOLDEN_NORM equal NONE will
leave the input coefficients unmodified.
It is strongly recommended to use spherical basis set when using the NWChem Molden output for JANPA analysis
Example input file for a scf calculation. The resulting Molden file will
be named h2o.molden
start heat
geometry; he 0. 0. 0.; end
basis spherical; * library 6-31g ; end
task scf
property
vectors heat.movecs
moldenfile
molden_norm janpa
end
task scf property
Then, the resulting h2o.molden file can be post processed by Janpa with the following command
java -jar janpa.jar h2o.molden > h2o.janpa.txt
Localization¶
Localized molecular orbitals (LMOs) can be computed with the localization keyword.
property
localization (( pm || boys || ibo) default pm) ((0 || 1 || 2) default 0)
end
The following methods are available:
An optional integer switch may be supplied after the localization method specification. The default 0 means only occupied orbitals are localized. Use 1 to localize virtual orbitals instead, or 2 to separately localize occupied and virtual orbitals in the same run. Localization of virtual orbitals is not implemented for the Boys localization. If only the keyword localization is given, this defaults to a Pipek-Mezey localization of the occupied molecular orbitals.
In spin-unrestricted calculations, alpha and beta spin orbitals are localized separately. Fractional-occupation DFT and restricted open-shell Hartree-Fock or Kohn-Sham are not yet supported; attempting to use localization with fractional orbital occupations may run but will likely produce unwanted and wrong results.
A localization run prints a characterizations of the LMOs. Specifically, the PM and IBO localizations print an energy-sorted list of LMOs indicating the density weight of each LMO on the various atoms for weights exceeding a 1% threshold; the atoms are listed in terms of their input number. A Boys localization prints the LMO centroid and the expectation value of r^2 per LMO.
Typically, however, the user will want to visualize the LMOs. The localization run saves the LMO coefficients in a file locorb.movecs, which can be used as the MO vector file in an NWChem DPLOT task to generate corresponding volume data maps, for example, in the `cube’ format commonly used in computational chemistry. The latter can be visualized with a considerable number of open source, free, or proprietary software packages.
NWChem test jobs that use the orbital localization functionality (under $NWCHEM_TOP/QA/tests/) are localize-ibo-allyl, localize-pm-allyl, and localize-ibo-aa. The latter is based on the acrylic acid example from the original IBO publication.17 IBO number 19 represents the delocalized C-C pi bond LMO of the system. After running the test job, the corresponding cube file for LMO 19 can be generated, for example, with
dplot
vectors locorb.movecs
limitxyz
-4.3334997 4.9318898 50
-3.3065879000000002 3.718637 50
-2.0 2.0 50
gaussian
spin alpha
orbitals view;1;19
output localize-ibo-aa-00019.cube
end
task dplot
The resulting plot with ±0.03 isosurfaces may look like the following
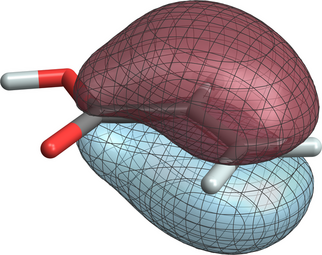
A Python script is available to facilitate the creation of volume data files for visualizing multiple MOs or LMOs of a given molecule. For the acrylic acid example, after running NWChem with the localize-ibo-aa.nw input, the following command will create an input file with dplot tasks to generate cubes for the 19 occupied and 10 virtual IBOs of the molecule on a 50x50x50 grid:
dplot.py -i localize-ibo-aa.nw -m locorb.movecs -g 50 -l 1-29
The dplot task shown above is included in the resulting NWChem input file dplot.nw.
Gaussian Cube Files¶
Electrostatic potential (keyword esp) and the magnitude of the
electric field (keyword efield) on the grid can be generated in the
form of the Gaussian Cube File. This behavior is triggered by the
inclusion of grid keyword as shown
below
grid [pad dx [dy dz]] [rmax x y z] [rmin x y z] [ngrid nx [ny nz]] [output filename]
where
-
pad dx [dy dz]- specifies amount of padding (in angstroms) in x,y, and z dimensions that will be applied in the automatic construction of the rectangular grid volume based on the geometry of the system. If only one number is provided then the same amount of padding will be applied in all dimensions. The default setting is 4 angstrom padding in all dimensions. -
rmin x y z- specifies the coordinates (in angstroms) of the minimum corner of the rectangular grid volume. This will override any padding in this direction. -
rmax x y z- specifies the coordinates (in angstroms) of the maximum corner of the rectangular grid volume. This will override any padding in this direction. -
ngrid nx [ny nz]- specifies number of grid points along each dimension. If only one number is provided then the same number of grid points are assumed all dimensions. In the absence of this directive the number of grid points would be computed such that grid spacing will be close to 0.2 angstrom, but not exceeding 50 grid points in either dimension. -
output filename- specifies name of the output cube file. The default behavior is to use prefix-elp.cubeor prefix-elf.cubefile names for electrostatic potential or electric field respectively. Here prefix denotes the system name as specified in start directive. Note that Gaussian cube files will be written in the run directory (where the input file resides).
Example input file
echo
start nacl
geometry nocenter noautoz noautosym
Na -0.00000000 0.00000000 -0.70428494
Cl 0.00000000 -0.00000000 1.70428494
end
basis
* library 6-31g*
end
#electric field would be written out to nacl.elf.cube file
#with
#ngrid : 20 20 20
#rmax : 4.000 4.000 5.704
#rmin :-4.000 -4.000 -4.704
property
efield
grid pad 4.0 ngrid 20
end
task dft property
#electrostatic potential would be written to esp-pad.cube file
# with the same parameters as above
property
esp
grid pad 4.0 ngrid 20 output esp-pad.cube
end
task dft property
#illustrating explicit specification of minumum box coordinates
property
esp
grid pad 4.0 rmax 4.000 4.000 5.704 ngrid 20
end
task dft property
References¶
-
Autschbach, J.; Patchkovskii, S.; Pritchard, B. Calculation of Hyperfine Tensors and Paramagnetic NMR Shifts Using the Relativistic Zeroth-Order Regular Approximation and Density Functional Theory. Journal of Chemical Theory and Computation 2011, 7 (7), 2175–2188. https://doi.org/10.1021/ct200143w. ↩
-
Aquino, F.; Pritchard, B.; Autschbach, J. Scalar Relativistic Computations and Localized Orbital Analyses of Nuclear Hyperfine Coupling and Paramagnetic NMR Chemical Shifts. Journal of Chemical Theory and Computation 2012, 8 (2), 598–609. https://doi.org/10.1021/ct2008507. ↩
-
Aquino, F.; Govind, N.; Autschbach, J. Scalar Relativistic Computations of Nuclear Magnetic Shielding and <i>g</i>-Shifts with the Zeroth-Order Regular Approximation and Range-Separated Hybrid Density Functionals. Journal of Chemical Theory and Computation 2011, 7 (10), 3278–3292. https://doi.org/10.1021/ct200408j. ↩
-
Aquino, F.; Govind, N.; Autschbach, J. Electric Field Gradients Calculated from Two-Component Hybrid Density Functional Theory Including Spin−orbit Coupling. Journal of Chemical Theory and Computation 2010, 6 (9), 2669–2686. https://doi.org/10.1021/ct1002847. ↩
-
Autschbach. Computation of Optical Rotation Using Timedependent Density Functional Theory. Computing Letters 2007, 3 (2), 131–150. https://doi.org/10.1163/157404007782913327. ↩
-
Autschbach, J. Time-Dependent Density Functional Theory for Calculating Origin-Independent Optical Rotation and Rotatory Strength Tensors. ChemPhysChem 2011, 12 (17), 3224–3235. https://doi.org/10.1002/cphc.201100225. ↩↩↩
-
Krykunov, M.; Autschbach, J. Calculation of Optical Rotation with Time-Periodic Magnetic-Field-Dependent Basis Functions in Approximate Time-Dependent Density-Functional Theory. The Journal of Chemical Physics 2005, 123 (11), 114103. https://doi.org/10.1063/1.2032428. ↩
-
Moore, B.; Srebro, M.; Autschbach, J. Analysis of Optical Activity in Terms of Bonds and Lone-Pairs: The Exceptionally Large Optical Rotation of Norbornenone. Journal of Chemical Theory and Computation 2012, 8 (11), 4336–4346. https://doi.org/10.1021/ct300839y. ↩↩
-
Krykunov, M.; Kundrat, M. D.; Autschbach, J. Calculation of Circular Dichroism Spectra from Optical Rotatory Dispersion, and Vice Versa, as Complementary Tools for Theoretical Studies of Optical Activity Using Time-Dependent Density Functional Theory. The Journal of Chemical Physics 2006, 125 (19), 194110. https://doi.org/10.1063/1.2363372. ↩
-
Hammond, J. R.; Govind, N.; Kowalski, K.; Autschbach, J.; Xantheas, S. S. Accurate Dipole Polarizabilities for Water Clusters n=212 at the Coupled-Cluster Level of Theory and Benchmarking of Various Density Functionals. The Journal of Chemical Physics 2009, 131 (21), 214103. https://doi.org/10.1063/1.3263604. ↩
-
Mullin, J. M.; Autschbach, J.; Schatz, G. C. Time-Dependent Density Functional Methods for Surface Enhanced Raman Scattering (SERS) Studies. Computational and Theoretical Chemistry 2012, 987, 32–41. https://doi.org/10.1016/j.comptc.2011.08.027. ↩
-
Aquino, F. W.; Schatz, G. C. Time-Dependent Density Functional Methods for Raman Spectra in Open-Shell Systems. The Journal of Physical Chemistry A 2014, 118 (2), 517–525. https://doi.org/10.1021/jp411039m. ↩
-
Fischer, S. A.; Ueltschi, T. W.; El-Khoury, P. Z.; Mifflin, A. L.; Hess, W. P.; Wang, H.-F.; Cramer, C. J.; Govind, N. Infrared and Raman Spectroscopy from Ab Initio Molecular Dynamics and Static Normal Mode Analysis: The C-H Region of DMSO as a Case Study. The Journal of Physical Chemistry B 2015, 120 (8), 1429–1436. https://doi.org/10.1021/acs.jpcb.5b03323. ↩
-
Aprà, E.; Bhattarai, A.; Baxter, E.; Wang, S.; Johnson, G. E.; Govind, N.; El-Khoury, P. Z. Simplified Ab Initio Molecular Dynamics-Based Raman Spectral Simulations. Applied Spectroscopy 2020, 74 (11), 1350–1357. https://doi.org/10.1177/0003702820923392. ↩
-
Pipek, J.; Mezey, P. G. A Fast Intrinsic Localization Procedure Applicable for Ab Initio and Semiempirical Linear Combination of Atomic Orbital Wave Functions. The Journal of Chemical Physics 1989, 90 (9), 4916–4926. https://doi.org/10.1063/1.456588. ↩
-
Foster, J. M.; Boys, S. F. Canonical Configurational Interaction Procedure. Reviews of Modern Physics 1960, 32 (2), 300–302. https://doi.org/10.1103/revmodphys.32.300. ↩
-
Knizia, G. Intrinsic Atomic Orbitals: An Unbiased Bridge Between Quantum Theory and Chemical Concepts. Journal of Chemical Theory and Computation 2013, 9 (11), 4834–4843. https://doi.org/10.1021/ct400687b. ↩↩
-
Knizia, G.; Klein, J. E. M. N. Electron Flow in Reaction Mechanisms-Revealed from First Principles. Angewandte Chemie International Edition 2015, 54 (18), 5518–5522. https://doi.org/10.1002/anie.201410637. ↩